脊髓性肌萎缩症SMA致病机制
脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA)是一种严重致死、致残性神经肌肉疾病。作为一种常染色体隐性遗传病,SMA致病基因SMN1定位于5q13.2,其编码的SMN蛋白是真核细胞生物生存所必需的管家蛋白,并维持运动神经元的存活。SMN1双等位基因发生致病性变异通常导致SMA的发生。
SMA突变基因类型主要有两大类:
1)95%的SMA由SMN1纯合缺失所致,即[0+0]基因型;
2)5%的SMA由复合杂合突变所致,即一个等位基因缺失,另一个等位基因发生微小致病性变异,为[0+1d]基因型。
此外,SMN1双等位基因均为微小致病性变异,即[1d+1d]基因型,非常罕见,目前仅有白种人近亲婚配的报道。
1891年,奥地利神经病学家Guido Werdnig,描述了最严重的I型SMA症状,这是SMA首次被发现和报道。可是它的遗传起因一直不详。直到1995年,法国研究人员在5号染色体上发现了一个基因, 叫做SMN1。随后,SMA的分子致病机制也逐渐被揭开:SMN1基因突变或者缺失,是造成SMA的主要原因。SMA的致病机理可定位于SMN1基因7号外显子(和/或8号外显子)的纯合缺失或复合杂合缺失。当然,在研究的过程中,一些疑问也接踵而来,特别是SMN1上的关键致病位置是7号外显子还是8号外显子,或是两个外显子都是,众说纷纭,我们引经据典,通过SMA指南、共识、权威文献等材料来逐步阐明外显子7和外显子8对于SMA发病的影响,供参考:
1. SMN1外显子7尾端存在一个终止密码子,外显子8改变与否,不影响SMN蛋白。
1)1995年《Structure and Organization of the Human Survival Motor Neurone (SMN) Gene》一文报道了SMN1基因的结构。SMN1外显子7尾端存在一个终止密码子,外显子8不参与SMN蛋白最终合成。由此可见,外显子8 改变与否,SMN蛋白编码不受影响。

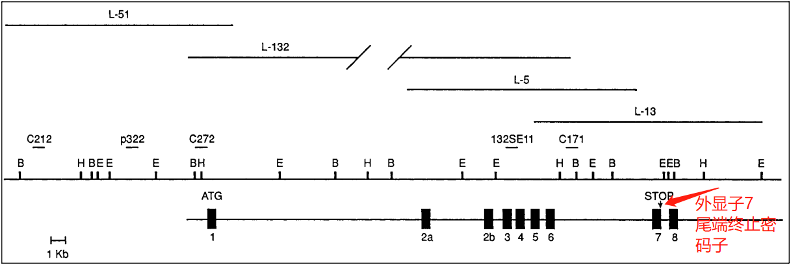
2)2015年《Newborn screening for spinal muscular atrophy:Anticipating an imminent need》一文,报道SMN1外显子7尾端存在一个终止密码子,在SMN1成熟的mRNA中不含外显子8。


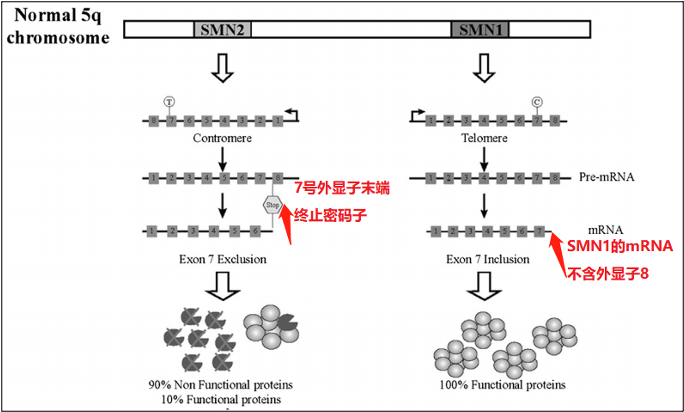
2. SMA指南及共识指出:SMN1外显子8位于非编码区
1)2020年3月《脊髓性肌萎缩症的临床实践指南》指出:SMN1第8外显子位于非编码区。

2)2020年11月《脊髓性肌萎缩症遗传学诊断专家共识》,指出:SMN1第8外显子位于非编码区,SMN1缺失通常指外显子7缺失。

由此,不难发现,从外显子8在SMN1基因上的位置可见,SMN1基因编码的SMN蛋白不包含外显子8,无论外显子8是否发生改变,均不会影响SMN蛋白的功能,因此,外显子8可以说与SMA的发生无关。
同时,由上述证据,我们也可知,外显子7可单独发生改变,与外显子8并不是完全协同改变的,因此,不能由外显子8是否存在,来间接判断外显子7的真实情况。
【参考文献】
1.L Bürglen, et al. "Structure and organization of the human survival motor neurone (SMN) gene. " Genomics 32.3(1996):479-82.
2.Phan, Han C et al. “Newborn screening for spinal muscular atrophy: Anticipating an imminent need.” Seminars in perinatology vol. 39,3 (2015): 217-29. doi:10.1053/j.semperi.2015.03.006
3.中华医学会医学遗传学分会遗传病临床实践指南撰写组. 脊髓性肌萎缩症的临床实践指南[J]. 中华医学遗传学杂志,2020,37(3):263-268.
4.北京医学会医学遗传学分会,北京罕见病诊疗与保障学会. 脊髓性肌萎缩症遗传学诊断专家共识[J]. 中华医学杂志,2020,100(40):3130-3140.